Since new types of neuronal ceroid lipofuscinoses (NCL) are constantly being discovered, new strategies to improve the diagnosis have become ever more important. A so-called "diagnostic strategy" is outlined in the following graphical depiction.
The steps toward posing the final diagnosis must differ according to the age of the patient and the symptoms observed. Different strategies apply for the congenital, infantile and late infantile NCLs, and NCL at a juvenile age of onset. Please note that the graphic only depicts NCL types that have an identifiable gene: The adult forms remain obscure.
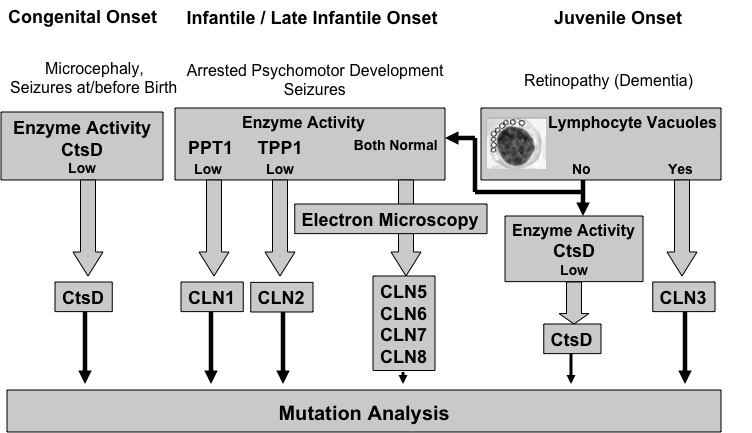
Figure 1: Schematic outline of the "Diagnostic Strategy" according to type of NCL
Congenital Onset of Disease
In cases of children who present with seizures at or even before birth and microcephaly, a rare congenital form of NCL should be considered. In those cases, enzyme activity of Cathepsin D (CtsD) must be measured.
At the University Medical Centre in Hamburg, Germany, in the Metabolic Diagnostics Laboratory of the Department of Paediatrics, we measure Cathepsin D activity in cultured fibroblasts derived from a tiny skin biopsy sample of the patient. A method for measuring Cathepsin D activity in blood samples is being established as well.
Referring physicians who would like to send us samples for Cathepsin D analysis should contact us (see Contact). We offer to send them all material and tubes that are needed for taking and sending the skin biopsy.
In case of low or missing Cathepsin D activity we demand the confirmation of this diagnosis by means of mutation analysis of the corresponding CtsD gene.
For mutation analysis 5-10 ml of so-called "EDTA blood" from the patient must be supplied and, if possible, from the parents as well (please consult your referring physician):
All samples for Cathepsin D analysis should be sent to the following address:
Metabolic Diagnostics Laboratory
Zoltan Lukacs, PhD / Angela Schulz, M.D.
Department of Paediatrics
Building N 23
University Medical Center Hamburg-Eppendorf
Martinistrasse 52
D-20246 Hamburg
Germany
Phone: +49-40-42803-3710
Fax: +49-40-42803-5137
Email: an.schulz@uke.uni-hamburg.de
info@ncl-netz.de
Infantile / Late Infantile Onset of Disease
In cases of infantile or late-infantile disease onset, enzyme activities of Palmitoyl-protein thioesterase I (PPT1 / CLN1-gene product) and Tripeptidyl-peptidase I (TPP1 / CLN2-gene product) must be measured.
At the University Medical Centre in Hamburg, Germany, in the Metabolic Diagnostics Laboratory of the Department of Paediatrics, a technique has been developed for measuring PPT1 and TPP1 enzyme activities in a few drops of blood. Referring physicians utilize a so-called “Guthrie Card” for this measurement.

Figure 2: A depiction of the so-called "Guthrie Card" for analysing dried blood spots (DBS) to determine PPT1 and TPP1 enzyme activity.
Guthrie cards with dried bloodspots can easily be mailed to our laboratory for enzyme measurement.
The blood should be dried correctly and sent in a normal envelope at room temperature to the following address (Please Note: Do NOT cover the blood samples with aluminium or plastic foil):
Metabolic Diagnostics Laboratory
Zoltan Lukacs, PhD / Angela Schulz, M.D.
Department of Paediatrics
Building N 23
University Medical Center Hamburg-Eppendorf
Martinistrasse 52
D-20246 Hamburg
Germany
Phone: +49-40-42803-3710
Fax: +49-40-42803-5137
Email: an.schulz@uke.uni-hamburg.de
info@ncl-netz.de
If PPT1 or TPP1 enzyme activity levels are low, or missing altogether, an infantile NCL (CLN1) or late infantile NCL (CLN2) can be diagnosed.
We demand the confirmation of this diagnosis by means of mutation analysis of the corresponding CLN1 or CLN2 gene. Such a mutation analysis can be performed at our Institute for Human Genetics. If the diagnosis has been confirmed there, a genetic counselling for patients and families is always offered.
For mutation analysis 5-10 ml of so-called "EDTA blood" from the patient must be supplied and, if possible, from the parents as well (please consult your referring physician):
Samples should be sent to the following address:
Professor Dr. Andreas Gal
Institute for Human Genetics
University Medical Center Hamburg-Eppendorf
Butenfeld 42
D-22529 Hamburg
Germany
A Normal PPT1 and TPP1 enzyme activity does not rule out NCL!
In all such cases we recommend examination by electron microscopy of lymphocytes or of a tiny skin biopsy sample from the patient.
The presence of lysosomal storage material as demonstrated by electron microscopy confirms the diagnosis of NCL. Examination of the specific ultrastructure of the storage material can help to distinguish between the rare NCL types in infantile or late infantile age.. Once the NCL diagnosis has been confirmed in one of these rare NCL types through the finding of lysosomal storage material, the final diagnostic step is also mutation analysis of other known NCL genes such as CLN5, CLN6 or CLN8. The decision as which gene should be analyzed first can be difficult since these NCL-forms are rare. We provide mutation analysis of CLN5, CLN6 and CLN8 at the University Medical Centre Hamburg, Germany. We coordinate electron microscopy and genetic analysis for these rare NCL-forms.
For further information please contact us (see Contact).
 back to top
back to top
Juvenile Onset of Disease
If the age at disease onset is "juvenile" ( about 5-8 years old), a blood smear with a few drops of fresh blood should be examined for lymphocyte vacuoles. The presence of vacuolized lymphocytes in combination with typical clinical NCL symptoms leads to the diagnosis of juvenile NCL.
We always demand confirmation of this diagnosis by means of mutation analysis of the corresponding CLN3 gene. Such a mutation analysis can be performed at our Institute for Human Genetics. If the diagnosis has been confirmed there, a genetic counselling for patients and families is always offered.
For mutation analysis 5-10 ml of so-called "EDTA blood" from the patient must be supplied and, if possible, from the parents as well (please consult your referring physician):
Samples should be sent to the following address:
Professor Dr. Andreas Gal
Institute for Human Genetics
University Medical Center Hamburg-Eppendorf
Butenfeld 42
D-22529 Hamburg
Germany
If no mutation in the CLN3-gene is found an NCL can still NOT be ruled out!. Some milder mutations in the CLN1- or CLN2-gene may cause the clinical signs of juvenile NCL. Other rare NCL-types such as Northern Epilepsy with Mental Retardation (EPMR, mutation of the CLN8-gene) or the recently discovered CLN9 NCL form can greatly resemble the classical juvenile NCL.
As in the infantile and late infantile NCL types, the presence of lysosomal storage material as demonstrated by electron microscopy confirms the NCL diagnosis. Examination of the specific ultrastructure of the storage material can help to distinguish between various non-classical NCL types. We offer to coordinate the electron microscopy and genetic diagnosis of these rare NCL types.
For further information please contact us (see Contact).
back to top
