
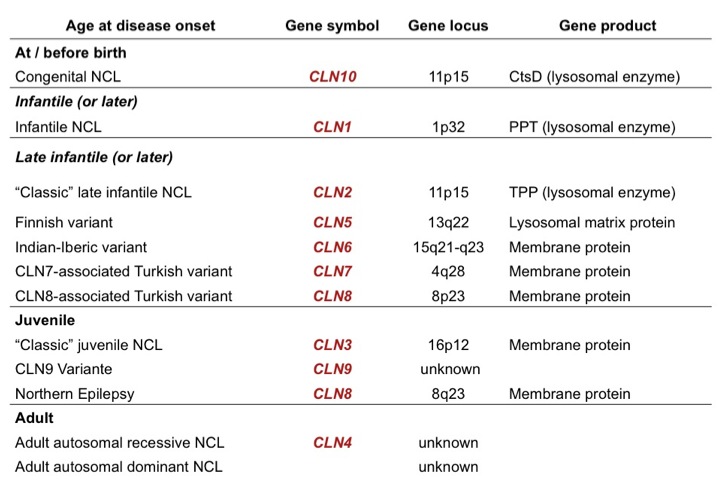
Inheritance: Autosomal-recessive
Age of Onset: 3 years
Genetics: -mutation of CLN2-gene (TPP1) (classic form)
-mutation of CLN5-gene (Finnish Variant, rare)
-mutation of CLN6-gene (Indian-Costa-Rican-Portuguese variant, rare)
-mutation of CLN8-gene (Turkish late-infantile variant, rare)
Clinical Picture: -progressice decline of psychomotor function
-visual failure caused by photoreceptor loss
-therapy-refractory epilepsy
-myoclonus
Diagnostics: -Tripeptidyl Peptidase1 (TPP1) enzyme defect
(Measurement of enzyme activity in dry blood spots, leucocytes, and skin fibroblasts)
-mutation analysis: CLN2-gene (frequent), CLN5, CLN6, CLN8 (rare)
-EM: intralysosomal storage material: curviliear bodies
DDx: -rare variants of late infantile NCL (CL5, CLN6, CLN7, CLN8)
Treatment: -palliative treatment only, no curative treatment available
-epilepsy: Valproic Acid, Lamotrigen are recommended
NOT recommended are Phenytoin, Vigabatrin
Juvenile NCL (JNCL / CLN3)
Inheritance: Autosomal-recessive
Age of Onset: 4-7 years
Genetics: -mutation of CLN3-gene (classic form)
-mutation of CLN1-gene (rare)
-mutation of CLN8-gene (Northern Epilepsy with Mental Retardation, EPMR)
-mutation of CLN9-gene (rare)
Clinical Picture: -progressive visual loss leading to complete amaurosis by age 10 years
-dementia
-epilepsy (Grand Mal)
-parkinsonism
-depression
-psychotic problems with hallucinations
Diagnostics: -blood smear: vacuolized lymphocytes
-mutation analysis: CLN3 (frequent), CLN8, CLN1 (rare)
-EM: intralysosomal storage material: fingerprint profiles, curviliear bodies
DDx: -Retinitis pigmentosa
-Hyperornithinemia
-other lysosomal disorders
-mitochondrial disorders
-peroxisomal disorders (M. Refsum)
-Nordic Epilepsy (no visual loss ) with CLN8 mutation
Treatment: -palliative treatment only, no curative treatment available
-epilepsy: Lamotrigen, Valproic Acid are recommended
NOT recommended are Carbamazepin, Phenytoin
-sleep disturbance, restlessness: Chloralhydrate
-psychotic symptoms with hallucinations:
-psychiatric consultation necessary
-medication: Levomepromazin, Clozapin, Risperidon
NCL Diagnostic Strategy
NCL Diagnostics
There is a growing number of different forms of neuronal ceroid-lipofuscinoses. This is one main reason why an economic strategy in NCL diagnostics has become very important.
The figure below will give you an overview of such a diagnostic strategy. It shows different diagnostic steps to be taken depending on the age of onset of NCL-symptoms.
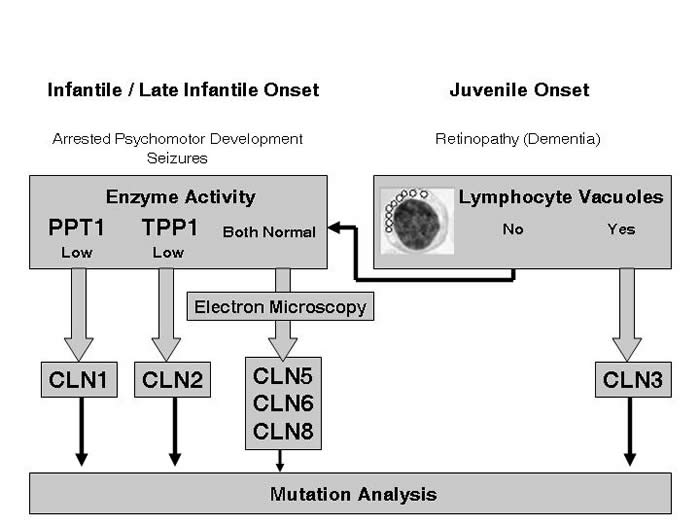
Figure 1: Overview Diagnostic Strategy for NCL Diseases
First of all the age of onset of the symptoms has to be determined.
Different diagnostic strategies apply to infantile / late infantile versus juvenile age of onset.
Infantile / Late
1) Infantile / Late Infantile Onset of Disease
If there is an infantile or late-infantile age of disease onset, enzyme activities of Palmitoyl-protein thioesterase I (PPT1 / CLN1-gene product) and Tripeptidyl-peptidase I (TPP1 / CLN2-gene product) should be measured. The metabolic diagnostics laboratory of the Department of Paediatrics at the University Medical Center Hamburg, Germany has established a technique to measure PPT1 and TPP1 enzyme activities in dry blood spots (see diagnostics).
Low or missing PPT1- or TPP1-enzyme activity leads ot the diagnosis of infantile NCL (CLN1) or late infantile NCL (CLN2).
We recommend to confirm this diagnosis by mutation analysis of the equivalent gene CLN1 or CLN2 respectively. This mutation analysis can be performed at our Institute for Human Genetics, Director Prof. Andreas Ga (see diagnostics). Once the diagnosis has been confirmed the Institute for Human Genetics offers genetic counseling for patients and families.
Normal PPT1- and TPP1-enzyme activity does not exclude a NCL-disease!
We recommend to examine lymphocytes or tissue from a skin biopsy by electron microscopy. The presence of lysosomal storage material demonstrated by electron microscopy confirms the diagnosis of NCL disease. Examination of the specific ultrastructure of the storage material can help to distinguish between rare NCL-forms with (late) infantile age of onset such as CLN6 or CLN8.
Once the diagnosis of NCL has been confirmed by the presence of lysosomal storage material the next diagnostic step is mutation analysis of other known NCL-genes like CLN5, CLN6 or CLN8.The decision which gene should be analyzed first can be difficult as these are rare NCL-forms. We provide mutation analysis of CLN5, CLN6 and CLN8 at the University Medical Center Hamburg, Germany, We also offer to coordinate electron microscopy and genetic diagnostics for these rare NCL-forms.
For further information please contact us (see Contact).
2) Juvenile Onset of Disease
If the age of disease onset is juvenile, a blood smear should be examined for lymphocyte vacuoles. The presence of vacuolized lymphocytes in combination with typical clinical NCL symptoms leads to the diagnosis of juvenile NCL.We recommend to confirm this diagnosis by mutation analysis of the equivalent gene CLN3 . This mutation analysis can be performed at our Institute for Human Genetics, Director Prof. Andreas Gal (see Contact). Once the diagnosis has been confirmed the Institute for Human Genetics offers genetic counseling for patients and families.
No mutation in the CLN3-gene will NOT exclude a NCL disease. Some milder mutations in the CLN1- or CLN2-gene may cause the clinical picture of juvenile NCL. Other rare NCL-forms like Northern Epilepsy with Mental Retardation (EPMR, mutation in the CLN8-gene) or the recently discovered new NCL-form CLN9 can present very similar to the classical juvenile NCL.Like for infantile and late infantile NCL-variants, the presence of lysosomal storage material demonstrated by electron microscopy confirms the diagnosis of NCL disease. Examination of the specific ultrastructure of the storage material can help to distinguish between different non-classical NCL-forms. We offer to coordinate electron microscopy and genetic diagnostics for these rare NCL-forms.
For further information please contact us (see Contact).